Julia H. Yang

Computational materials scientist working closely with experiments to address urgently-needed solutions for energy storage and the environment. Postdoc at Harvard University, Ph.D. from U.C. Berkeley.
I am an Environmental Fellow at Harvard University Center for the Environment (HUCE) working with the Materials Intelligence Research group since July 2022. Previously, I was a Ph.D. student at University of California, Berkeley with the Ceder group and an AI Resident with X (formerly Google X).
As a materials scientist working closely with experiments using computation and machine learning, my research elucidates thermodynamic and kinetic behavior of atoms or molecules to understand and design urgently-needed, sustainable materials for energy storage and the environment.
Please see Google Scholar for a full list of publications, my CV here, or my Faculty Candidate poster here.
Diversity, Equity, Inclusion, and Belonging
We are committed to fostering environments that are professionally and personally inclusive, equitable, psychologically safe, and welcoming to all teammates. We do this by leading with kindness, open-mindedness, and empathy.
- Kindness: We communicate openly and respectfully, remembering that it is okay for others to have different opinions from us. We treat others with the kindness we want others to treat us.
- Open-mindedness: We leave assumptions behind, and seek first to understand. We are aware of community biases and are persistent in finding new ways to reduce them. Lastly we actively encourage our peers to hold themselves to these same standards.
- Empathy: We always remember that we are on the same team, working together on science and engineering challenges that need our solutions. We give ourselves and others the time, space, and patience to grow and learn.
These are our guiding principles as ethical scientists and engineers.
Research interests & contributions
I. Atomistic resolution of electrodes
We are interested in the thermodynamics of battery electrode materials. We use computation to investigate intragranular and intergranular atomic-level features that limit electrochemical performance, such as rate capability and structural stability.
Previous work:
Using density functional theory (DFT) and random enumeration of large unit-cells (containing ~3000 atoms), we teamed up with an electrochemist and a microscopist to investigate the effect of Jahn-Teller-active iron in stabilizing a high-voltage, sodium-ion intercalation electrode. We suggest a lower bound for iron concentration to accommodate phase stability at high state of charge.
J. C. *. Kim, D.-H. *. Kwon, J. H. Yang *, H. Kim, S.-H. Bo, L. Wu, H. Kim, D.-H. Seo, T. Shi, J. Wang, Y. Zhu, and G. Ceder, “Direct Observation of Alternating Octahedral and Prismatic Sodium Layers in O3-Type Transition Metal Oxides”, Adv. Energy Mater., vol. 10, no. 31, p. 2 001 151, 2020. doi:10.1002/aenm.202001151.
*: equal contribution
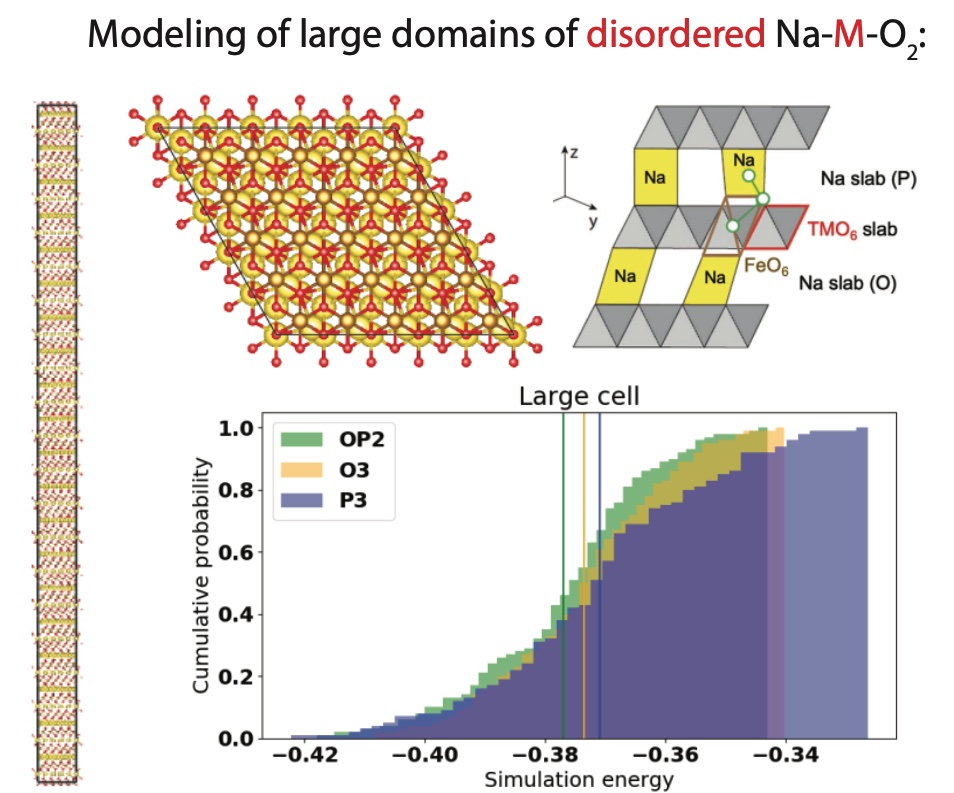
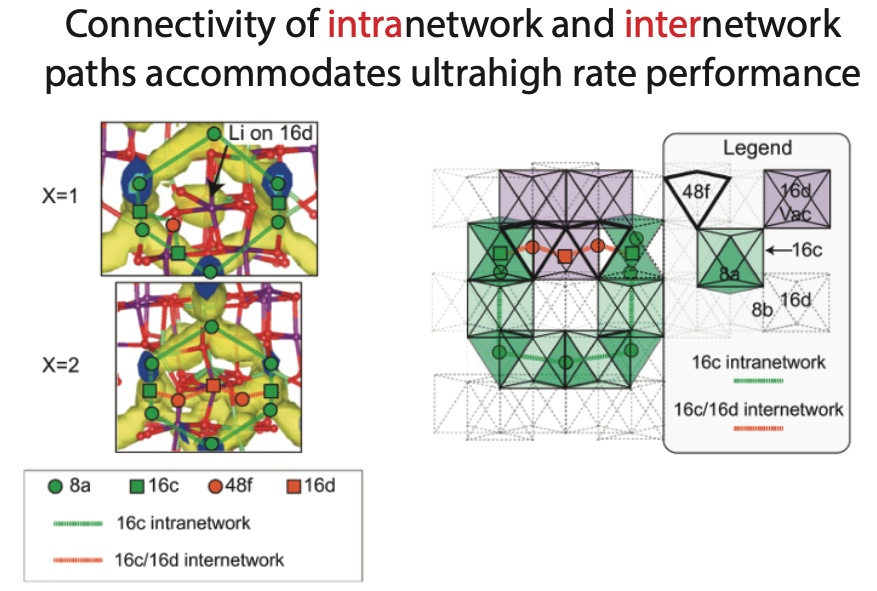
Random enumeration cannot capture experimentally-observed phenomena such as short-range cation order existing at the nano-domain level. We have also used DFT and lattice cluster expansion models to study a partially-disordered spinel positive electrode with ultrahigh power and energy density. Our model reveals that a novel internetwork pathway for Li traveling through newly-activated 48f-16d sites explains the origin of its high rate:
J. H. Yang† and G. Ceder†, “Activated internetwork pathways in partially-disordered spinel cathode materials with ultrahigh rate performance”, Adv. Energy Mater., vol. 13, no. 4, p. 2 202 955, 2023. doi:10.1002/aenm.202202955.
†: corresponding author
II. Computational methods: understanding and application
As computational materials scientists, we need to know the utilities and limitations of our approaches. For example, DFT methods are system-dependent and oftentimes cannot be used “out-of-the-box”. Understanding the applicability of our computational toolbox will be essential in studying complex electrochemical systems and under-explored interfaces.
Previous work:
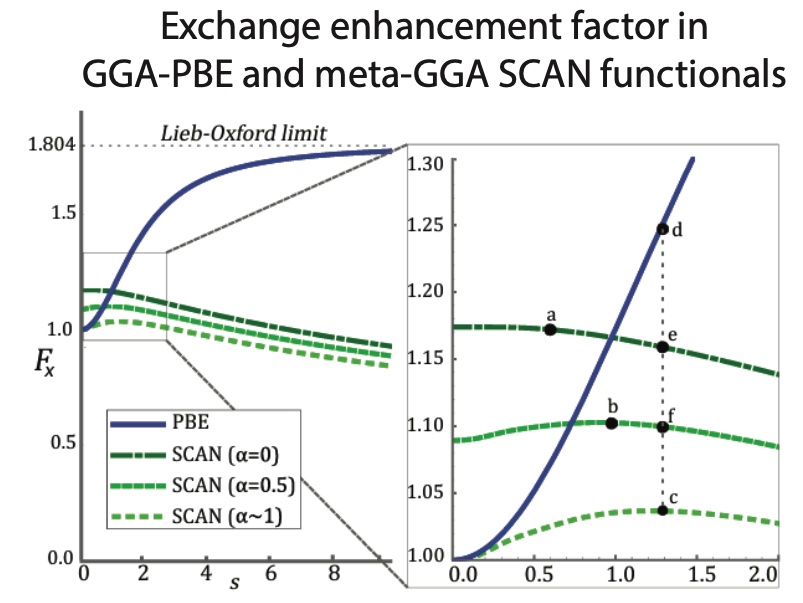
Given the efficacy of meta-GGA functionals in ground state crystal structure prediction, as observed in our work, we used data-driven methods to report systematic under-coordination in the well-known PBE functional, not observed in SCAN, and trace the origin of this difference to their exchange enhancement factors:
J. H. Yang, D. A. Kitchaev, and G. Ceder, “Rationalizing accurate structure prediction in the meta-GGA SCAN functional”, Phys. Rev. B, vol. 100, no. 3, p. 35 132, 2019. doi:10.1103/PhysRevB.100.035132.
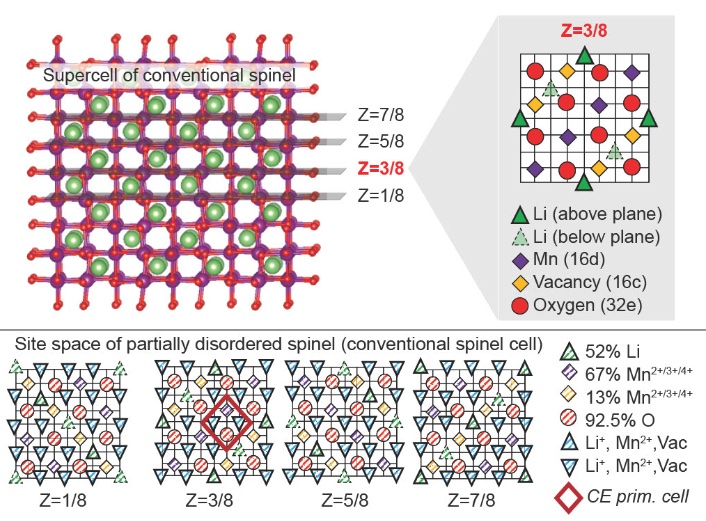
Lattice models can be used to model disordered systems, but they have rarely been extended to study systems with more than 2 or 3 species. In studying the partially-disordered spinel system, we constructed one of the most complex lattice models to our knowledge, and suggested that model error with CE models should scale with species complexity. We also explained our approaches in a “Behind the Paper” commentary:
J. H. Yang, T. Chen, L. Barroso-Luque, Z. Jadidi, and G. Ceder, “Approaches for handling high-dimensional cluster expansions of ionic systems”, npj Comput. Mater., vol. 8, no. 1, p. 133, 2022. doi: 10.1038/s41524-022-00818-3.
We are also the first to study the DFT corrections needed to model organic solvents. (Preprint coming soon.)
J. H. Yang†, A. W.-S. Ooi, Y. Xie, Z. A. Goodwin, J. Ding, S. Falletta, A.-H. A. Park, and B. Kozinsky†, “Thermal decomposition of the ethaline deep eutectic solvent”, in preparation.
III. Battery recycling with solvents
Molecular understanding of metal-solvent interactions are essential to develop new metals recycling methods. We are interested in exploiting the tunability of “designer solvents”, such as molten salts, ionic liquids, or deep eutectic solvents, to enable selective metal extraction.
Previous work:
We have developed a computational workflow, using cheminformatics and Bayesian methods, to navigate reaction conditions for optimizing chemical recycling of waste plastics. Several patents regarding this process have been filed by X Development LLC. See our primary contribution.
J. H. Yang, V. Gharakhanyan, T. Gadhiya, and A. Holiday, “Ionic liquid-based depolymerization optimization”, U.S. Patent App. 17/967,711, filed Oct. 17, 2022.
We also use large-scale atomistic simulations powered by state-of-the-art machine learning methods to understand molecular reactions in neat solvent. (Preprint coming soon.)
J. H. Yang†, A. W.-S. Ooi, Y. Xie, Z. A. Goodwin, J. Ding, S. Falletta, A.-H. A. Park, and B. Kozinsky†, “Thermal decomposition of the ethaline deep eutectic solvent”, in preparation.